PhyloProfile, (R)Tool for phylogenetic profiling
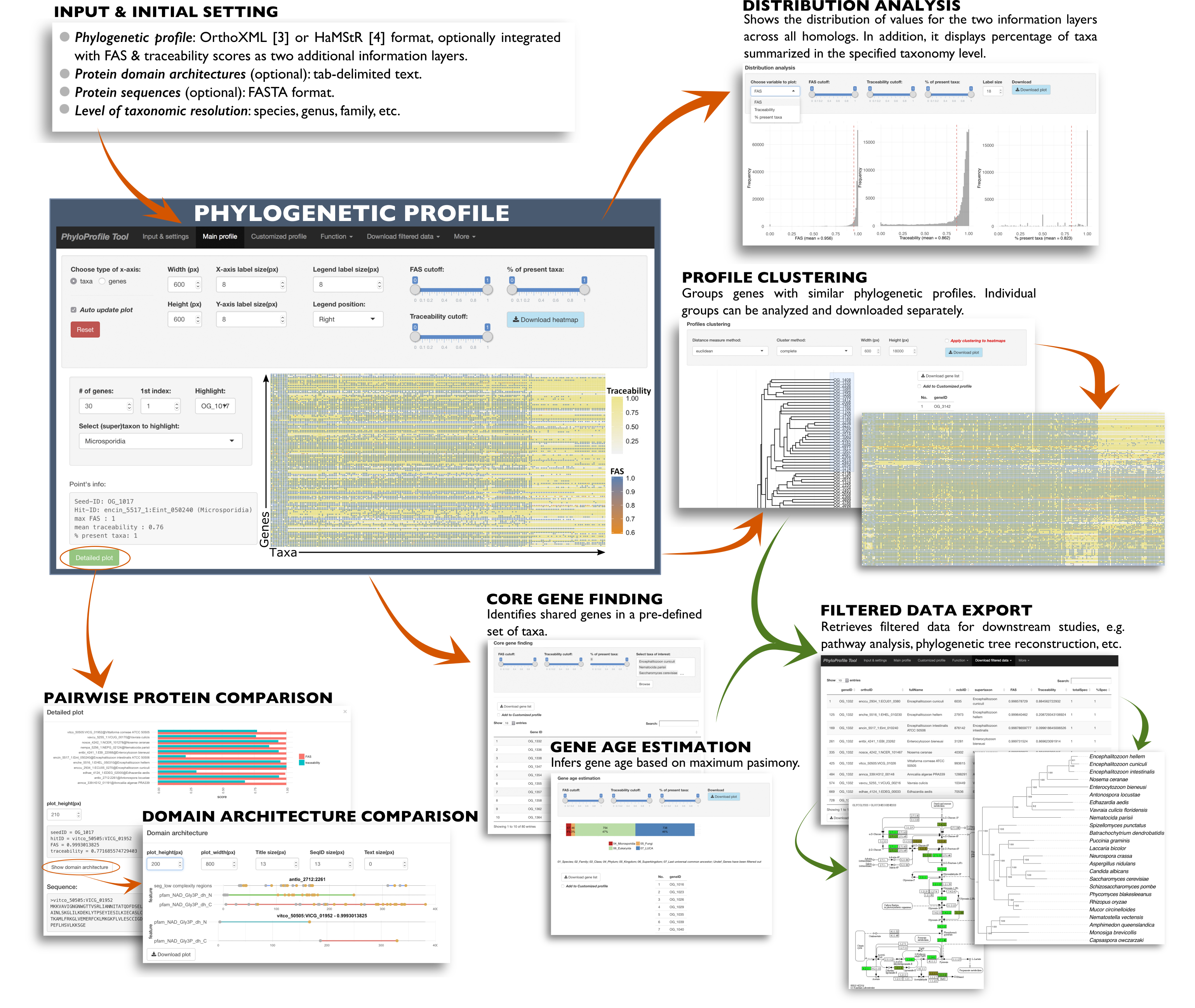
PhyloProfile is a Shiny-based tool for integrating, visualizing and exploring multi-layered phylogenetic profiles.
Alongside the presence/absence patterns of orthologs across large taxon collections, PhyloProfile allows the integration of any two additional information layers. These complementary data, like sequence similarity between orthologs, similarities in their domain architecture, or differences in functional annotations enable a more informed interpretation of phylogenetic profiles.
By utilizing the NCBI taxonomy, PhyloProfile can dynamically collapse taxa into higher systematic groups. This enables rapidly changing the resolution from the comparative analyses of proteins in individual species to that of entire kingdoms or even domains without changes to the input data.
PhyloProfile furthermore allows for a dynamic filtering of profiles – taking the taxonomic distribution and the additional information layers into account. This, along with functions to estimate the age of genes and core gene sets facilitates the exploration and analysis of large phylogenetic profiles.
PhyloProfile is based on the R-package Shiny, as such a recent version of R is needed. Once that is out of the way you can just clone this repository to get a copy of PhyloProfile.
git clone https://github.com/BIONF/phyloprofile
To start PhyloProfile simply move into the PhyloProfile directory and run the main script
cd PhyloProfile
Rscript phyloprofile.R
The initial start can take a while, as phyloprofile.R will try do download and install all necessary dependencies automatically. (Note: Depending on your system this sometimes fails, please check the console log for error messages concerning the dependency installation)
Once all packages are downloaded and installed your web browser will open a new tab and display the main PhyloProfile menu.
In data/demo/ you can find some test data to see how the files should look like. In our Wiki you can find a walkthrough through the different files and how they relate to the different functions of PhyloProfile.
Bugs
Any bug reports or comments, suggestions are highly appreciated. Please open an issue on GitHub or be in touch via email.
Acknowledgements
We would like to thank 1) Bastian for the great initial idea and his kind support, 2) Members of Ebersberger group for many valuable suggestions and …bug reports :)
Code of Conduct & Lisence
This tool is released with a Contributor Code of Conduct & under MIT lisence.