My first contribution to an open source software package

Yuppiiii!!! You know how you feel, when your work is getting recognized, right? ;)
Some time back, I was working on an issue with - yes, again - with PhyloProfile, where I need to sort a list of taxa based on their taxonomy distance. From Bastian - the guy who knows everything, I got to know taxize, a greate library from rOpenSci project for playing with NCBI taxonomy database. taxize has a function called class2tree() which create a tree object from a given list of species.
>library(taxize)
> spnames <- c('Homo_sapiens',
+ 'Pan_troglodytes',
+ 'Macaca_mulatta',
+ 'Mus_musculus',
+ 'Rattus_norvegicus',
+ 'Bos_taurus',
+ 'Canis_lupus',
+ 'Ornithorhynchus_anatinus',
+ 'Xenopus_tropicalis',
+ 'Takifugu_rubripes')
> out <- classification(spnames, db='ncbi')
> tr <- class2tree(out)
> plot(tr)
and this is the tree tr we got
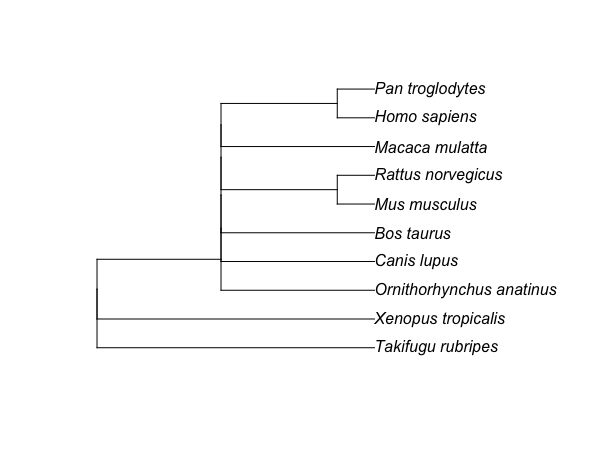
This tree has many unresolved splits, since class2tree() remove all unrank levels from the taxonomy ranks leading to a missing information for separating those splits (unrank levels are ranks that are named as “no rank” on the following table).
> out$Homo_sapiens
name rank id
1 cellular organisms no rank 131567
2 Eukaryota superkingdom 2759
3 Opisthokonta no rank 33154
4 Metazoa kingdom 33208
5 Eumetazoa no rank 6072
6 Bilateria no rank 33213
7 Deuterostomia no rank 33511
8 Chordata phylum 7711
9 Craniata subphylum 89593
10 Vertebrata no rank 7742
11 Gnathostomata no rank 7776
12 Teleostomi no rank 117570
13 Euteleostomi no rank 117571
14 Sarcopterygii no rank 8287
15 Dipnotetrapodomorpha no rank 1338369
16 Tetrapoda no rank 32523
17 Amniota no rank 32524
18 Mammalia class 40674
19 Theria no rank 32525
20 Eutheria no rank 9347
21 Boreoeutheria no rank 1437010
22 Euarchontoglires superorder 314146
23 Primates order 9443
24 Haplorrhini suborder 376913
25 Simiiformes infraorder 314293
26 Catarrhini parvorder 9526
27 Hominoidea superfamily 314295
28 Hominidae family 9604
29 Homininae subfamily 207598
30 Homo genus 9605
31 Homo sapiens species 9606
Bastian opened an issue in taxize github repository. At that time, I could already solve the issue (mostly) with my Perl code. But while writing the manuscript for PhyloProfile, we decided to convert the Perl code into R, so that we don’t have many scripts in many different languages in one program :-D
Eventually, I’ve not only improved the sorting result while implementing the algorithm in R (by using APE tree object), but I could also rewrite the class2tree() function to include the full taxonomy information for creating the species tree. By that I can sucessfully reconstruct the NCBI taxonomy tree, yahooo!!
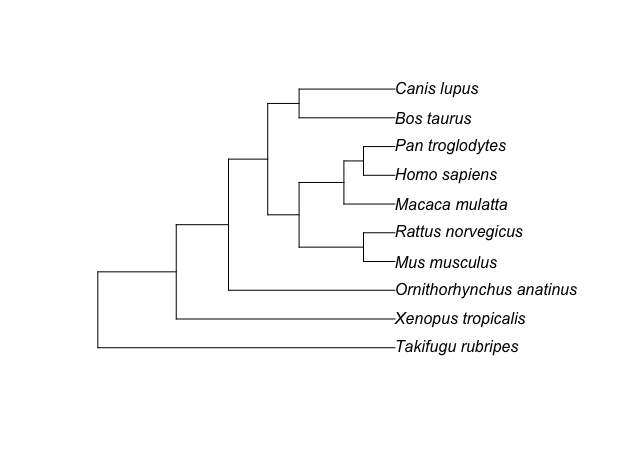
Bastian and I made a pull request in taxize. He helped me to run the tests they require. And finally, my code & our effort have been accepted ^_^
🎉 to @trvinh_ for his first contribution to an open source package! 🍾 https://t.co/PfGF22kvLy
— Bastian Greshake (@gedankenstuecke) 7. Oktober 2017
I have learned so many practical things with this first contribution. Thank you so much, Bastian ;-)